编辑推荐:
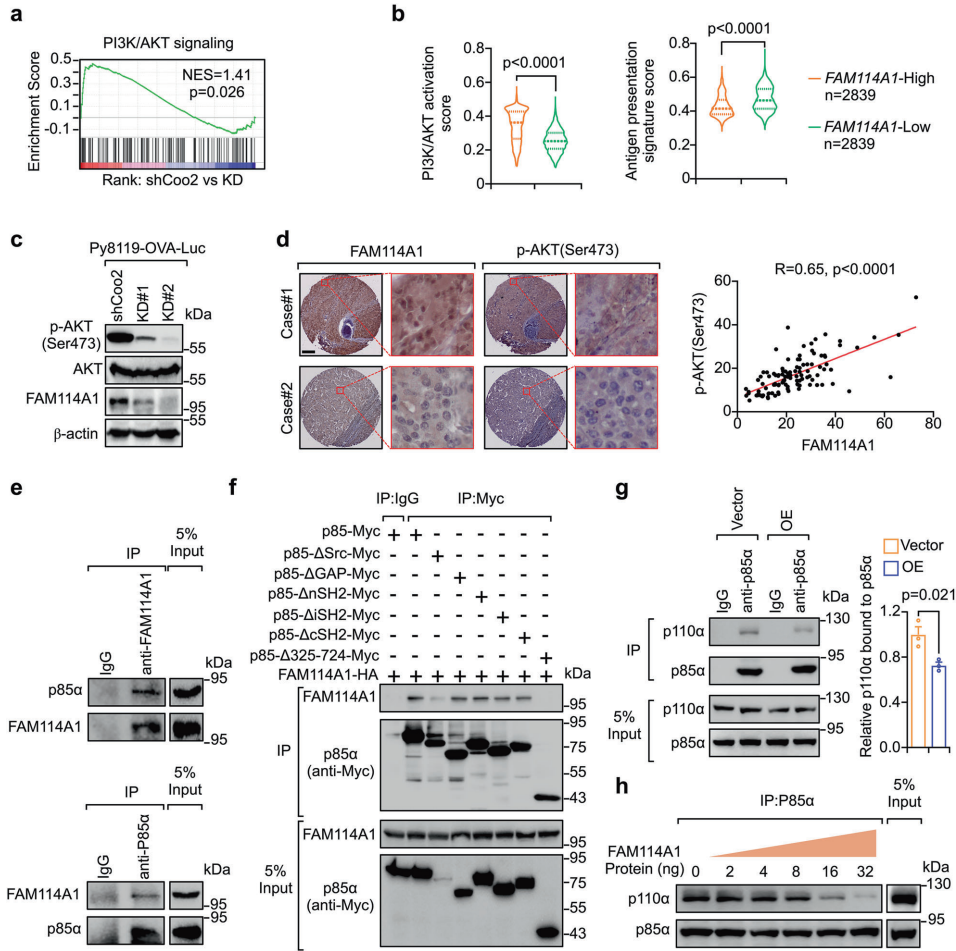
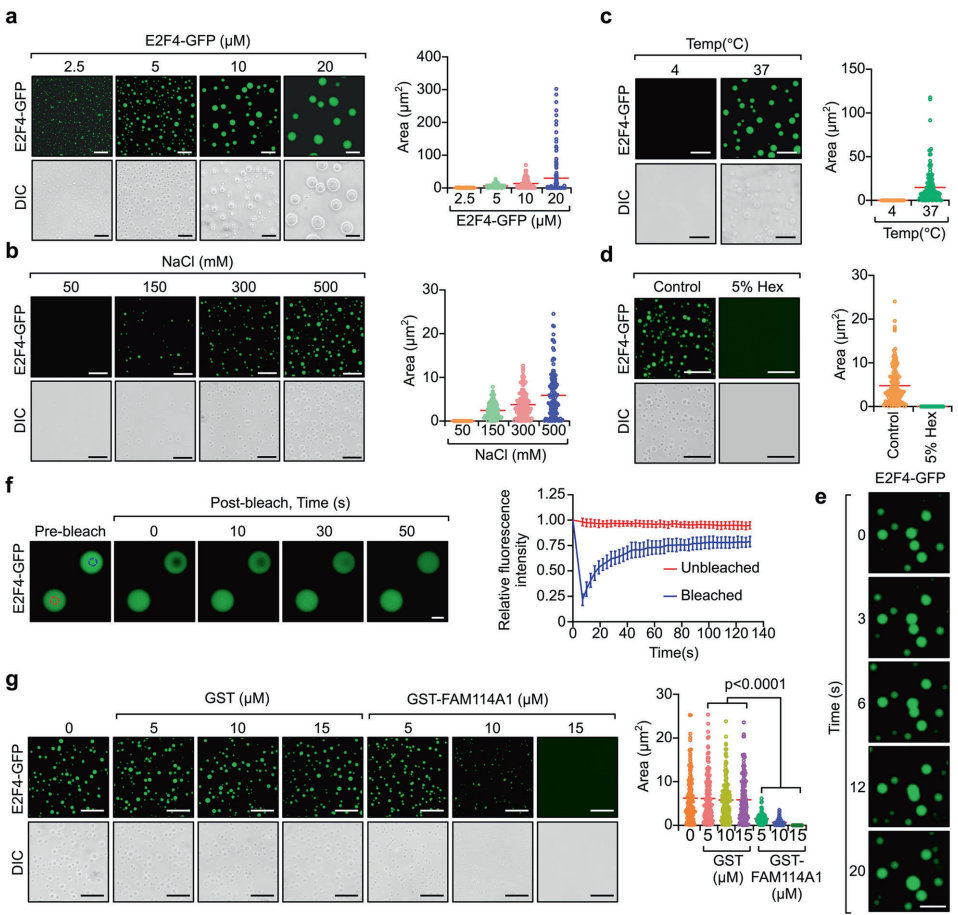
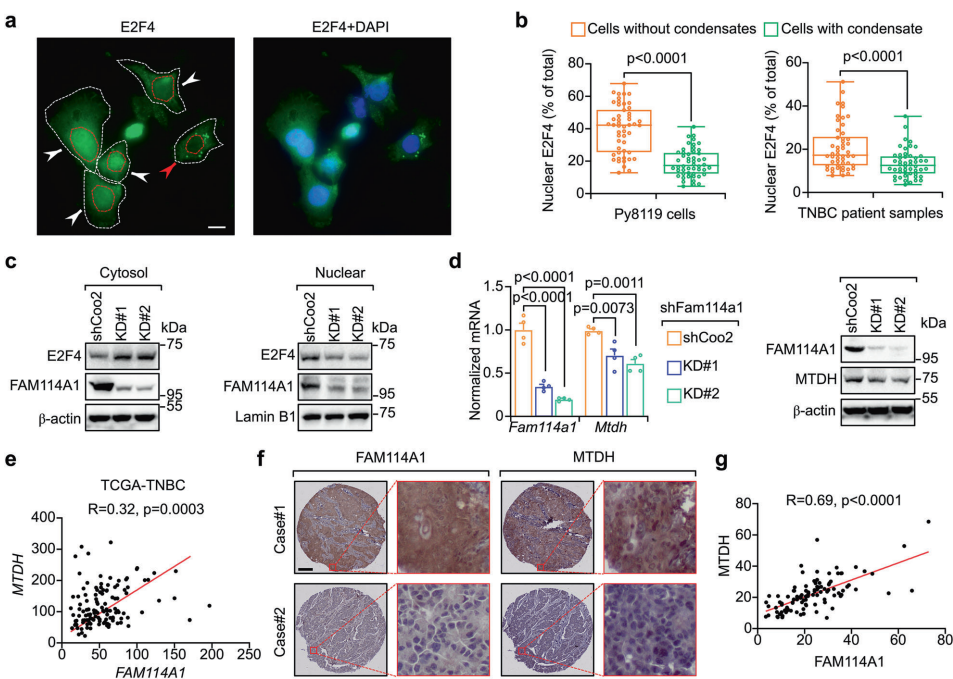
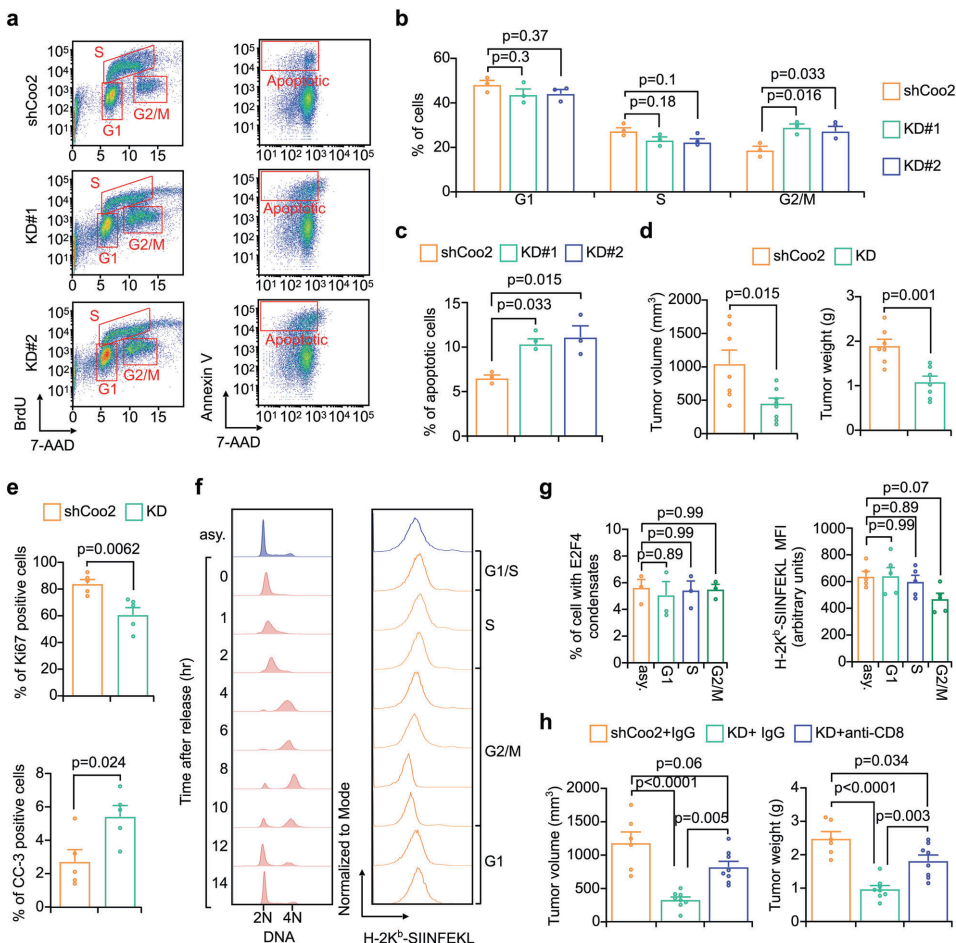
本研究针对三阴性乳腺癌(TNBC)免疫检查点阻断(ICB)疗法耐药性难题,通过CRISPR激活(CRISPRa)筛选发现FAM114A1是介导免疫逃逸的关键因子。机制上,FAM114A1通过结合p85α破坏p85α/p110α复合体激活PI3K/AKT通路,同时抑制E2F4转录因子液-液相分离(condensate formation),促进E2F4驱动的MTDH转录,双重作用抑制肿瘤抗原呈递。动物实验中靶向FAM114A1可增强抗PD-1疗效,临床队列分析显示FAM114A1特征能预测ICB应答。该研究为TNBC免疫治疗耐药提供了新的生物标志物和联合治疗策略。
生物通 版权所有





