编辑推荐:
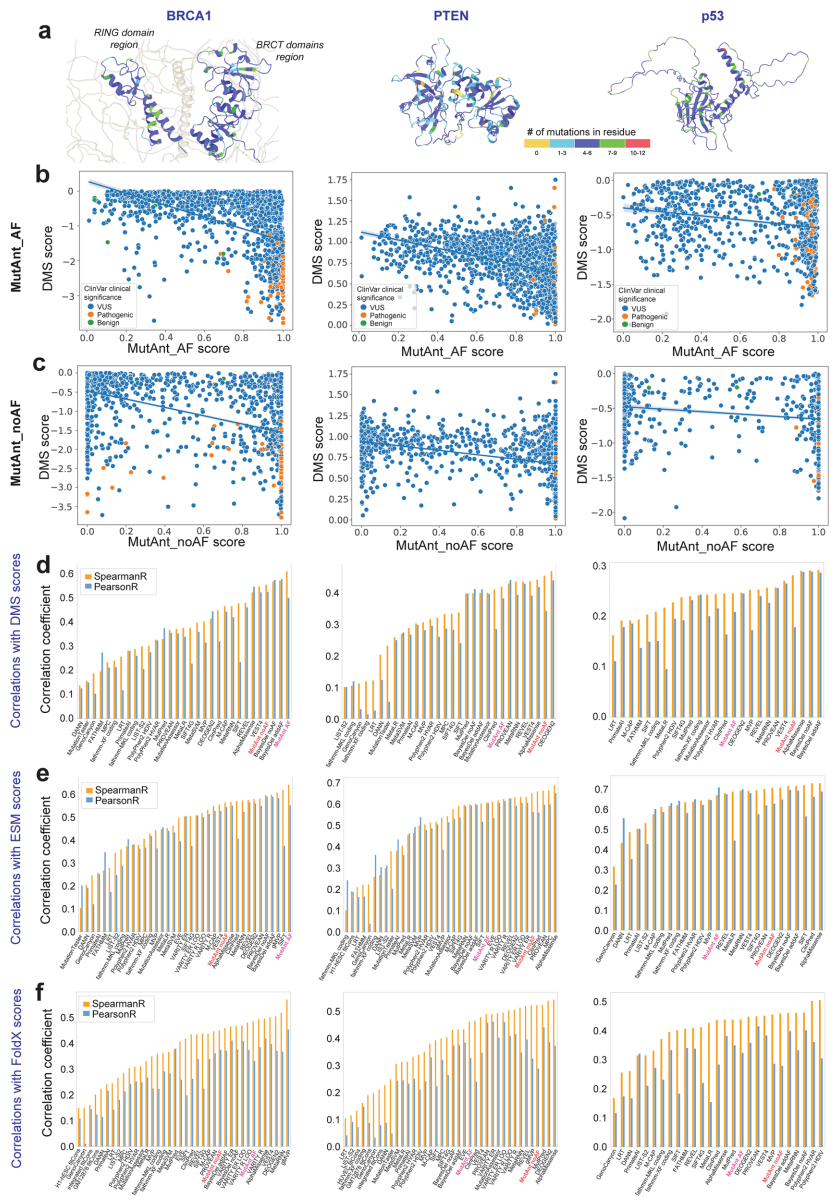
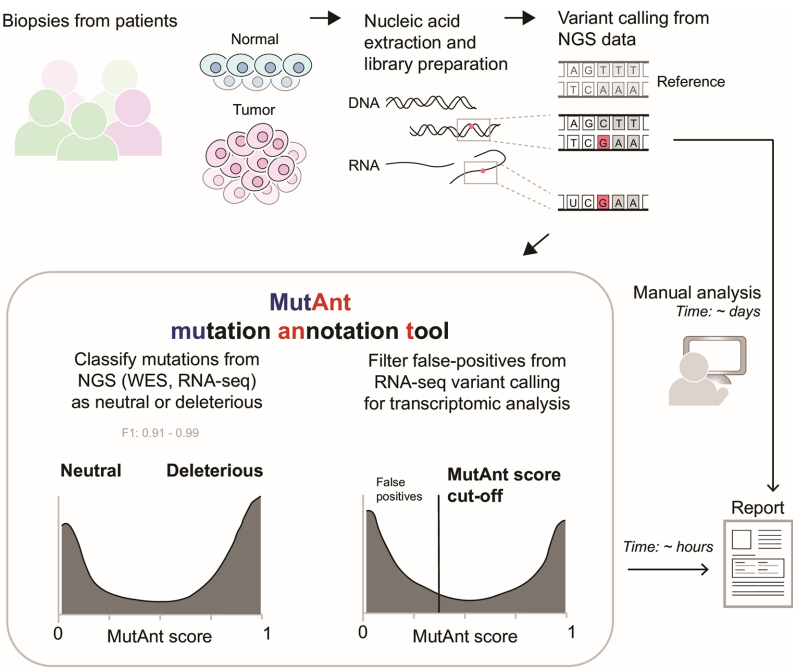
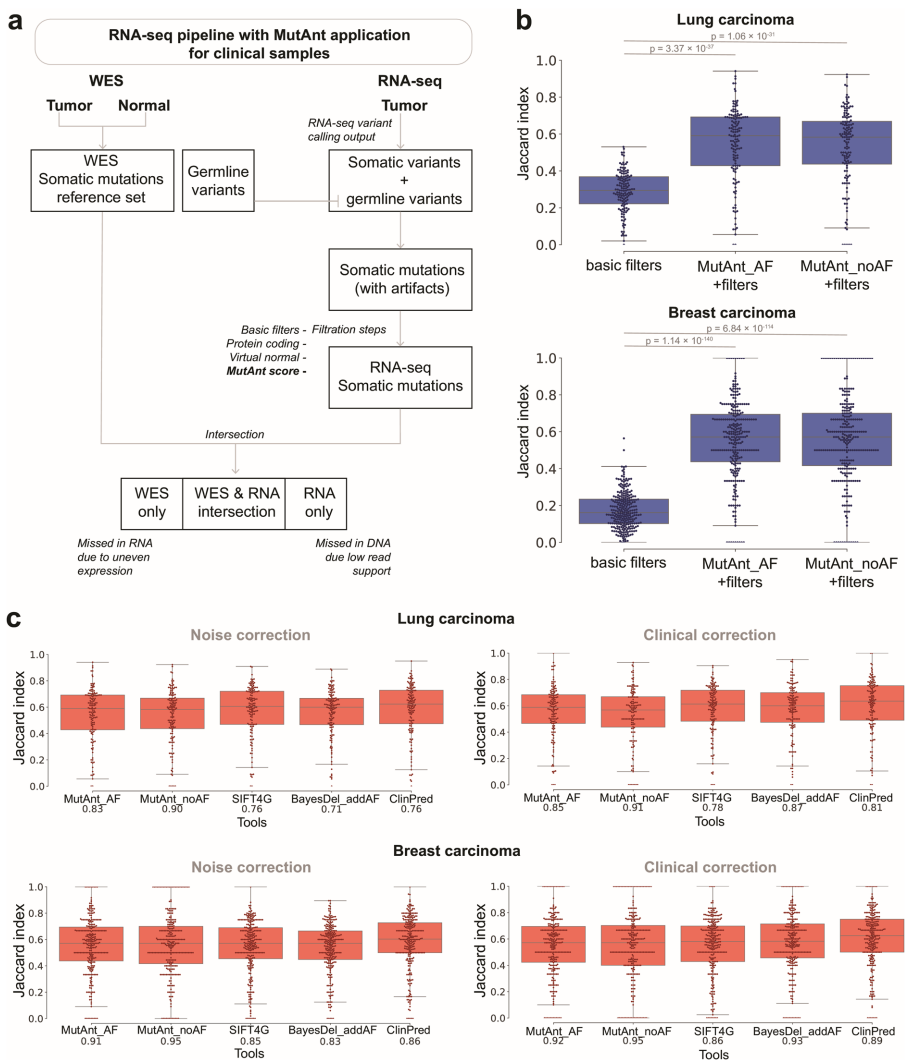
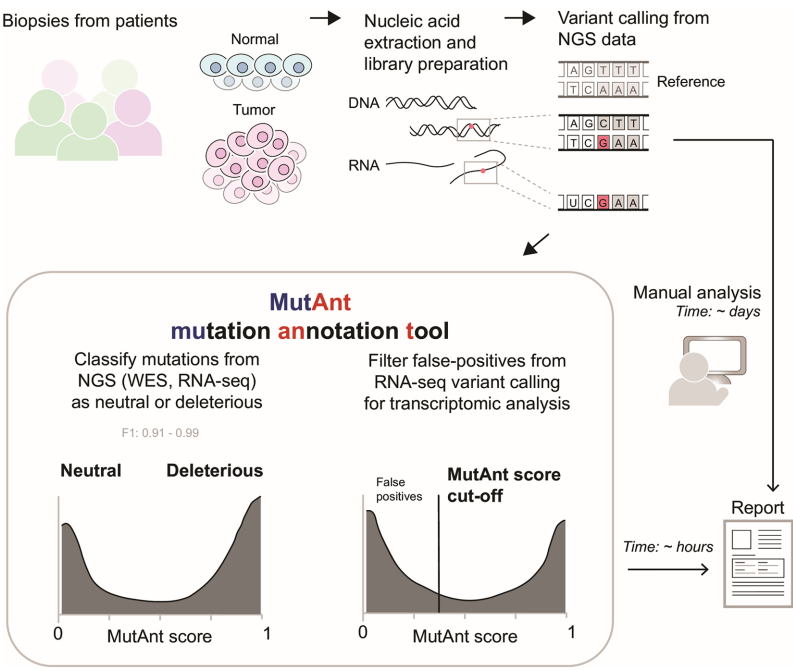
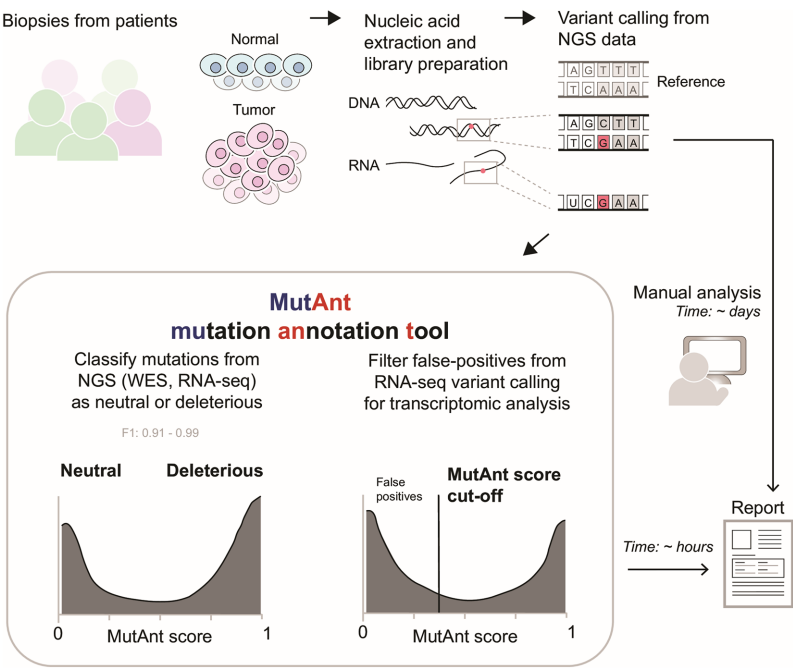
本研究针对罕见和新生变异致病性注释难题,开发了基于机器学习的突变元注释工具MutAnt。该工具整合群体等位基因频率、进化保守性评分及多算法预测结果,在独立验证集中表现出超高判别性能(F1-score: 0.88-0.99,ROC-AUC: 0.98-0.99),其有害性评分与BRCA1/PTEN/p53的深度突变扫描功能评分显著相关(ρ=0.28-0.61)。特别值得关注的是,应用MutAnt阈值过滤可显著提升RNA-seq体细胞变异检测的精准度,为临床基因组解读提供了可靠的计算支持。
生物通 版权所有





