编辑推荐:
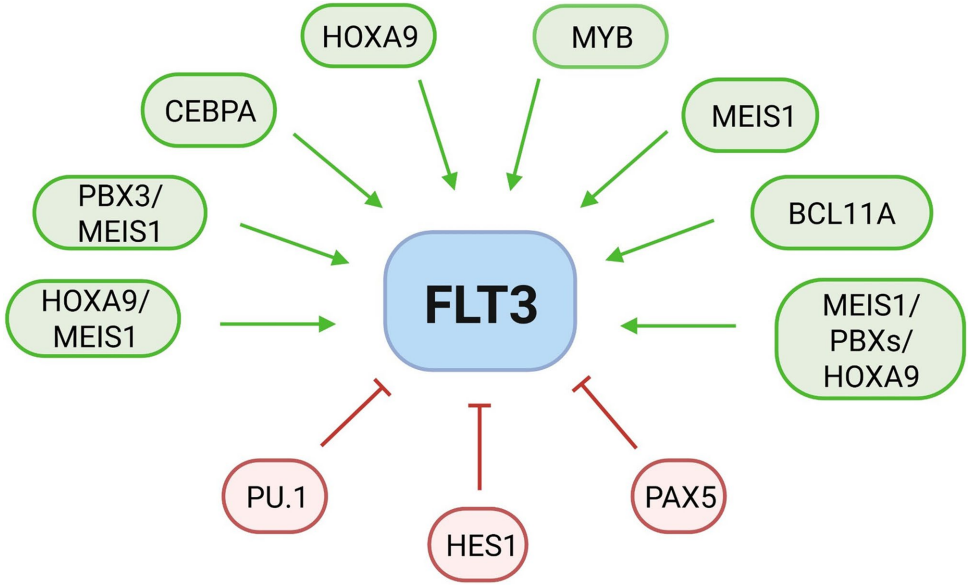
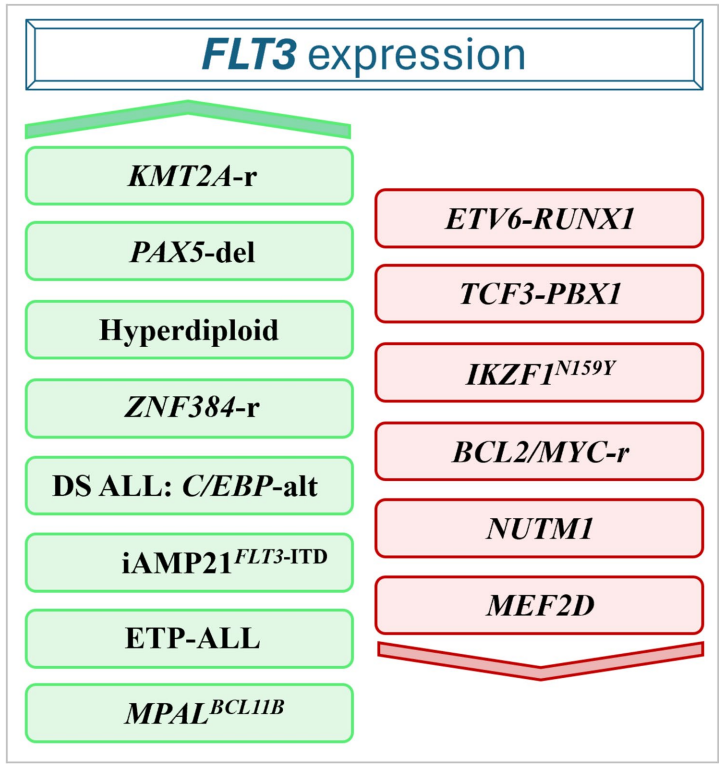
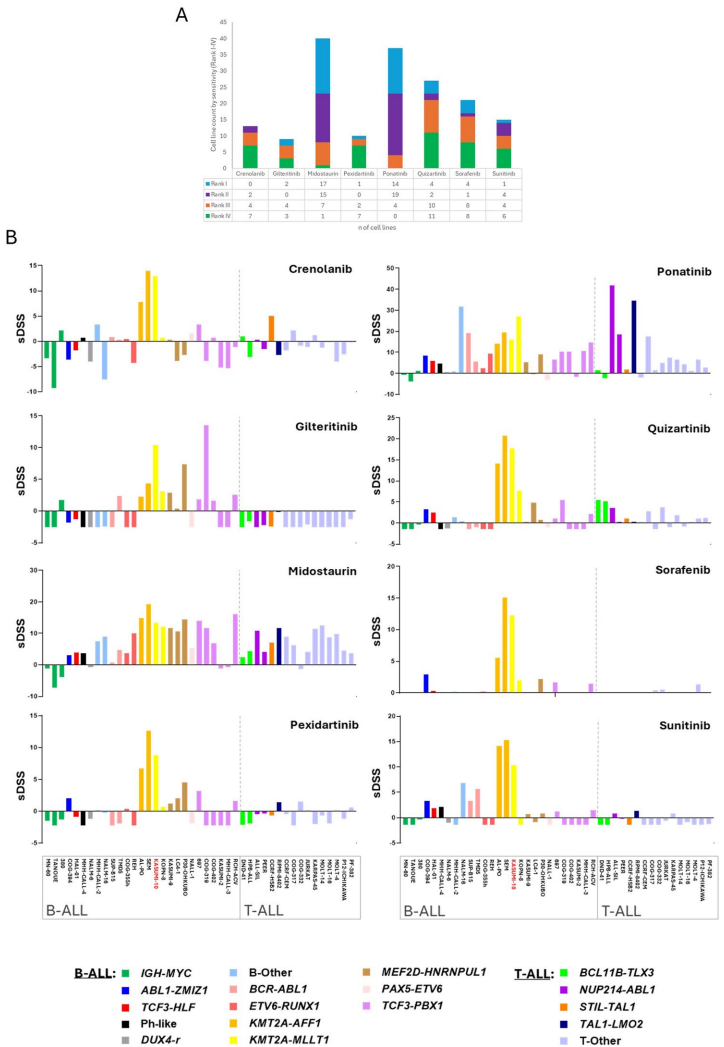
本综述系统阐述了FLT3(Fms样酪氨酸激酶3)在急性白血病中的最新研究进展。文章不仅总结了FLT3在急性髓系白血病(AML)中的经典突变(如ITD/TKD)及其靶向治疗经验,更着重揭示了其在急性淋巴细胞白血病(ALL)中独特的激活机制,包括非经典突变、表观遗传调控、环状RNA(circRNA)融合及三维基因组(3D基因组)改变等。作者探讨了基于FLT3抑制剂(FLT3i)、抗体及CAR-T等新兴疗法在ALL中的应用潜力,强调了将FLT3表达谱和全编码突变筛查纳入ALL(及AML)诊断,以实现精准医疗的迫切性。本文为FLT3作为泛白血病靶点的重新定义提供了有力证据。



 生物通微信公众号
生物通微信公众号
生物通 版权所有






