编辑推荐:
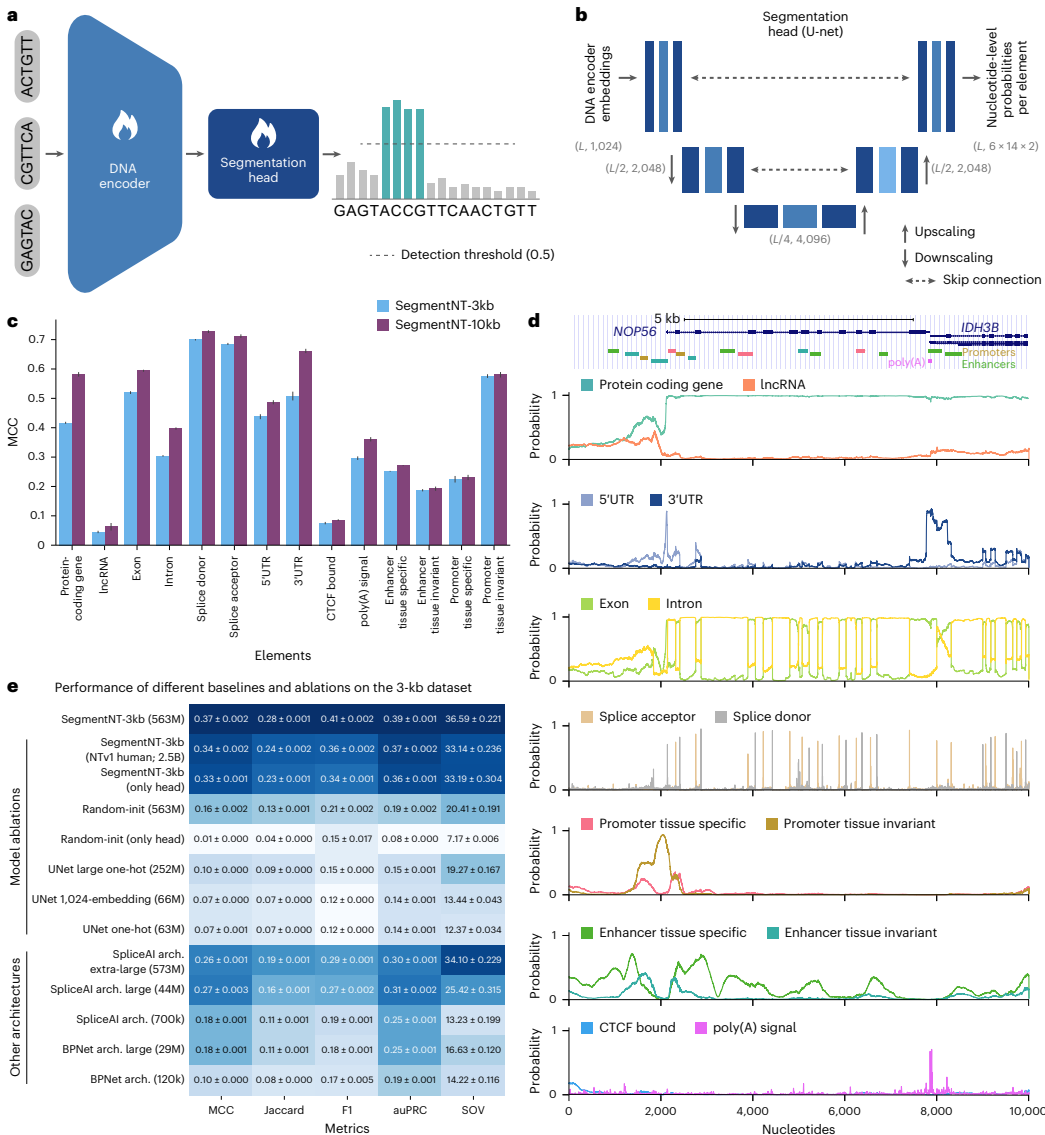
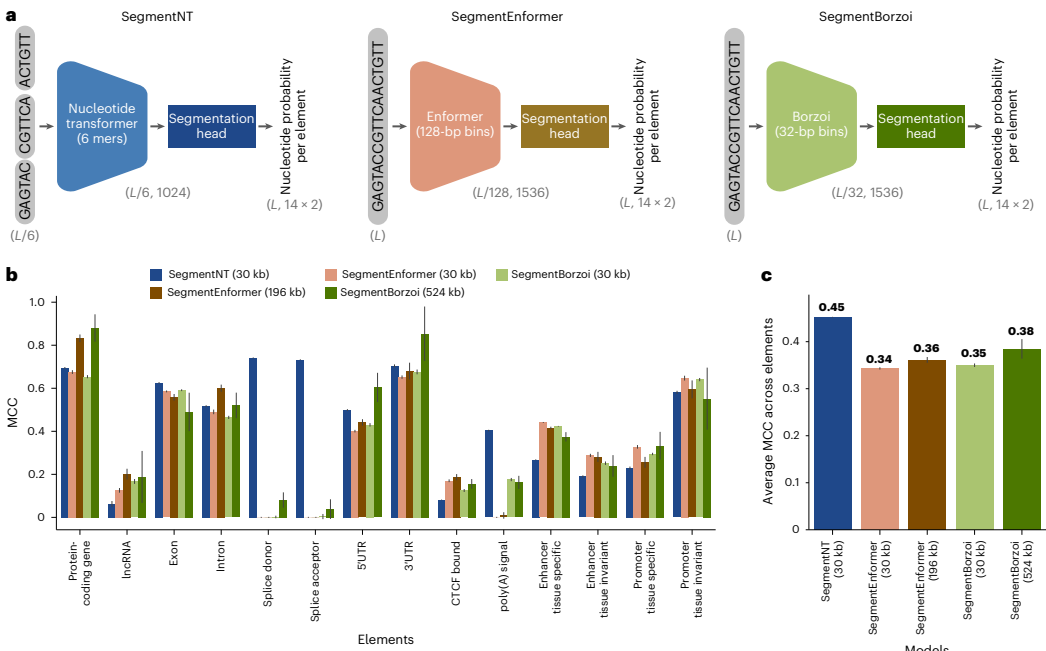
本研究针对当前基因组注释工具存在元素类别特异性强、训练数据有限等挑战,提出了基于预训练DNA基础模型的多标签语义分割方法SegmentNT。通过微调Nucleotide Transformer等模型,实现了14种基因和调控元件的单核苷酸分辨率精准定位,在50 kb长序列中达到最先进性能。该框架支持多物种泛化,为基因组注释提供了高效通用解决方案。
 生物通微信公众号
生物通微信公众号
生物通 版权所有






