编辑推荐:
本研究针对冷冻电子断层扫描(cryo-ET)技术中分子物种识别困难的瓶颈问题,开发了包含六种分子物种的实验性"幻影"数据集。研究人员通过整合细胞裂解液与纯化蛋白,建立了具有全面真实标注的标准化数据集,利用深度学习算法DeepFindET和CellCanvas实现了注释效率的十倍提升。该成果为算法开发提供了关键基准测试平台,相关数据已通过CryoET Data Portal开放共享,将显著推动原位结构生物学研究的发展。
细胞作为生命的基本单元,其内部复杂的分子机器如何协同工作一直是结构生物学的核心问题。冷冻电子断层扫描技术(cryo-ET)因其能在近天然状态下解析生物大分子三维结构的独特优势,已成为揭示细胞超微结构的重要工具。然而,这项技术面临着一个关键瓶颈——在低信噪比的断层扫描图像中,准确识别和标注各种分子物种仍然主要依赖人工操作,这不仅效率低下,也严重制约了高通量数据的解析效率。
传统基于模板匹配的标注方法存在明显局限性,而新兴的机器学习算法虽展现出潜力,却因缺乏标准化的大规模训练数据集而发展受阻。正是针对这一关键问题,由Ariana Peck和Yue Yu等来自Chan Zuckerberg Imaging Institute的研究团队在《Nature Methods》发表了这项开创性研究。他们精心设计了一个包含六种代表性分子物种的实验性"幻影"数据集,为算法开发提供了理想的基准测试平台。
研究人员采用的主要技术方法包括:1)通过功能化电镜网格捕获溶酶体富集的细胞裂解液,模拟真实细胞环境;2)整合六种分子量跨度达一个数量级的纯化蛋白(从268kDa的β-淀粉酶到4.3MDa的核糖体);3)使用Krios G4电镜收集1,070组倾斜系列图像;4)开发DenoisET算法进行图像降噪处理;5)结合PyTom模板匹配、二维板层挑选和人工标注生成初始标记;6)应用DeepFindET和CellCanvas深度学习模型实现自动化标注。
研究结果部分展示了系统的实验设计和严谨的数据验证:
"Creating and annotating a phantom dataset"详细描述了数据集的构建过程。研究人员巧妙地将溶酶体富集的HEK293T细胞裂解液与六种纯化蛋白混合,通过抗GFP纳米抗体捕获技术确保样品厚度约200nm,模拟典型细胞薄片条件。倾斜系列采集范围设定为±45°,像素尺寸1.51Å,总剂量控制在62.93e-/Å2。
"The CryoET Data Portal and related infrastructure"介绍了数据管理平台。研究团队建立了包含492张断层扫描图的标注数据集,其中训练集7张、公开测试集121张和私有测试集364张。所有数据采用OME-Zarr格式存储,可通过Neuroglancer浏览器或napari桌面查看器进行可视化分析。
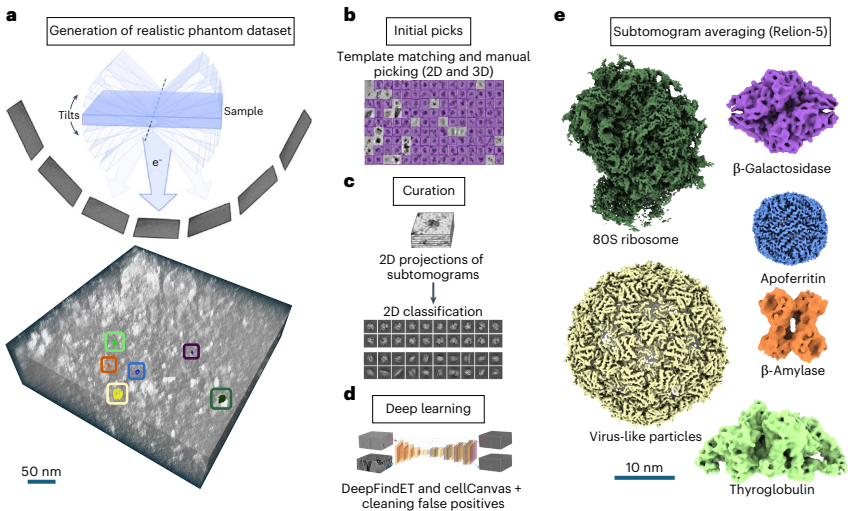
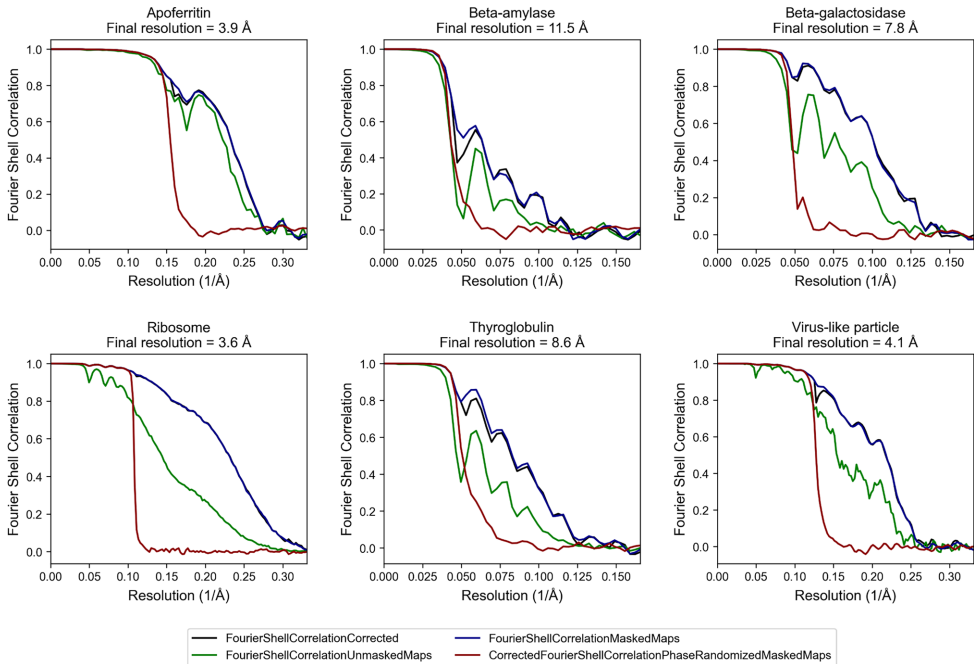
"Generating ground truth"部分详细阐述了标注流程的优化。通过结合三维模板匹配、二维板层挑选和人工标注,研究人员最终获得了约60,000个高质量标签。这些标签经过严格验证,各目标分子的三维重构分辨率达到:核糖体3.6Å(24,338颗粒)、病毒样颗粒4.1Å(3,022颗粒)、甲状腺球蛋白8.6Å(6,211颗粒)、β-半乳糖苷酶7.8Å(3,113颗粒)、脱铁铁蛋白3.9Å(23,393颗粒)和β-淀粉酶11.5Å(2,464颗粒)。
研究团队还开发了多项创新工具:DenoisET实现了基于Noise2Noise算法的断层扫描图降噪;copick建立了存储无关的标注平台;slab-picking开发了替代三维模板匹配的初始候选挑选方法;ArtiaX提供了断层扫描图中颗粒挑选的可视化工具;DeepFindET改进了原始DeepFinder包的卷积神经网络架构;CellCanvas实现了基于绘画的交互式分割。
这项研究的科学意义主要体现在三个方面:首先,标准化的幻影数据集解决了算法开发缺乏统一基准的关键问题,为机器学习方法在cryo-ET领域的应用奠定了基础。其次,研究展示的标注流程将人工参与从单颗粒水平提升到质量控制层面,显著提高了标注效率。最后,通过Kaggle竞赛验证,该数据集成功吸引了跨领域研究者的参与,最佳解决方案甚至超越了作者团队开发的DeepFindET模型,体现了开放科学的价值。
正如作者强调的,这项工作的最大价值在于建立了一个动态发展的资源平台。随着新算法的不断涌现,标注结果可以持续更新优化,最终形成更完善的真实标签集。这种开放、协作的研究模式,将为推动冷冻电子断层扫描技术的自动化分析带来深远影响,加速我们对于细胞分子机器运作机制的认知。
 生物通微信公众号
生物通微信公众号
生物通 版权所有






