编辑推荐:
本研究针对肥胖相关椎间盘退变(IVDD)的分子机制,揭示了饱和脂肪酸通过诱导线粒体损伤,释放mt-dsRNA激活PKR,进而促进NLRP3炎症小体介导的髓核细胞焦亡的新通路。研究人员通过体内外实验证实PKR缺失可缓解IVDD,并发现二甲双胍通过保护线粒体抑制该通路,为IVDD的防治提供了新靶点和治疗策略。
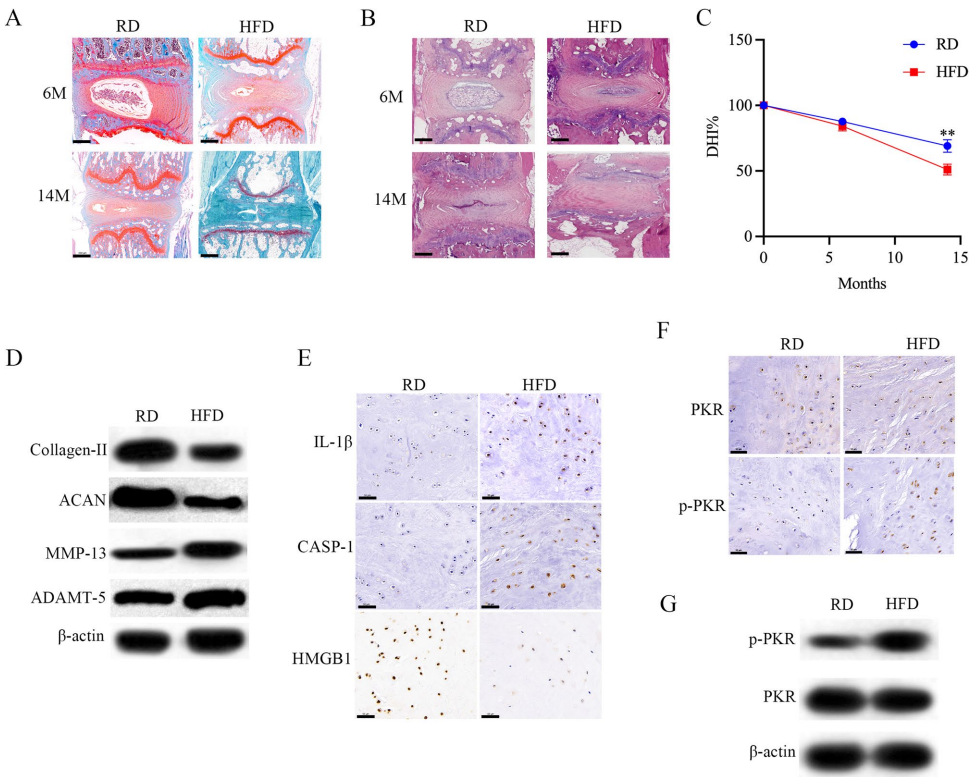
 p<0.05,p<0.001 by Student's t test,n=3,E intracellular HMGB1, visualized with red immunofluorescence staining, is predominately localized in the nucleus in NP cells. Following by exposure to palmitic acid,nuclear HMGB1 is released from the nucleus to the cytoplasm and knock off PKR significantly inhibited HMGB1 release. HMGB1(red), DAPI(blue), scale bar=50μm;F Caspase-1 activation and IL-1β cleavage were assessed by Western blot,n=3.PKR+/+ or PKR-/- NP cells were stimulated as indicated.G,H) PKR+/+ or PKR-/- NP cells were treated with palmitic acid as indicated; Supernatant was detected by ELISA. Cytotoxicity was determined by LDH assay,p<0.05,*p<0.001 by Student's t test,n=3,I the scanning electron microscopy showed that membrane pore-forming was increased by palmitic acid and PKR knock off decreased the membrane pore-forming in NP cells induced by palmitic acid; scale bar=20 um(below: magnification of the tetrago-num; scale bar=2μm).PA:palmitic acid'>
p<0.05,p<0.001 by Student's t test,n=3,E intracellular HMGB1, visualized with red immunofluorescence staining, is predominately localized in the nucleus in NP cells. Following by exposure to palmitic acid,nuclear HMGB1 is released from the nucleus to the cytoplasm and knock off PKR significantly inhibited HMGB1 release. HMGB1(red), DAPI(blue), scale bar=50μm;F Caspase-1 activation and IL-1β cleavage were assessed by Western blot,n=3.PKR+/+ or PKR-/- NP cells were stimulated as indicated.G,H) PKR+/+ or PKR-/- NP cells were treated with palmitic acid as indicated; Supernatant was detected by ELISA. Cytotoxicity was determined by LDH assay,p<0.05,*p<0.001 by Student's t test,n=3,I the scanning electron microscopy showed that membrane pore-forming was increased by palmitic acid and PKR knock off decreased the membrane pore-forming in NP cells induced by palmitic acid; scale bar=20 um(below: magnification of the tetrago-num; scale bar=2μm).PA:palmitic acid'>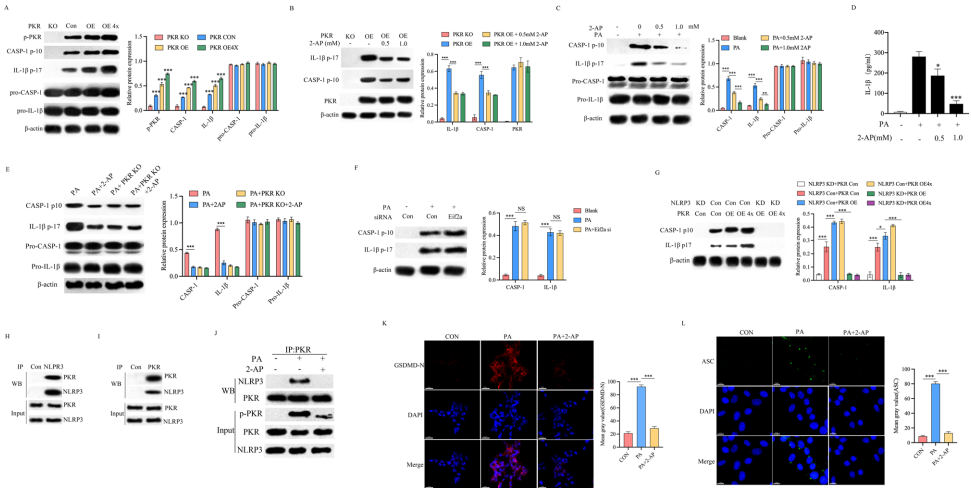
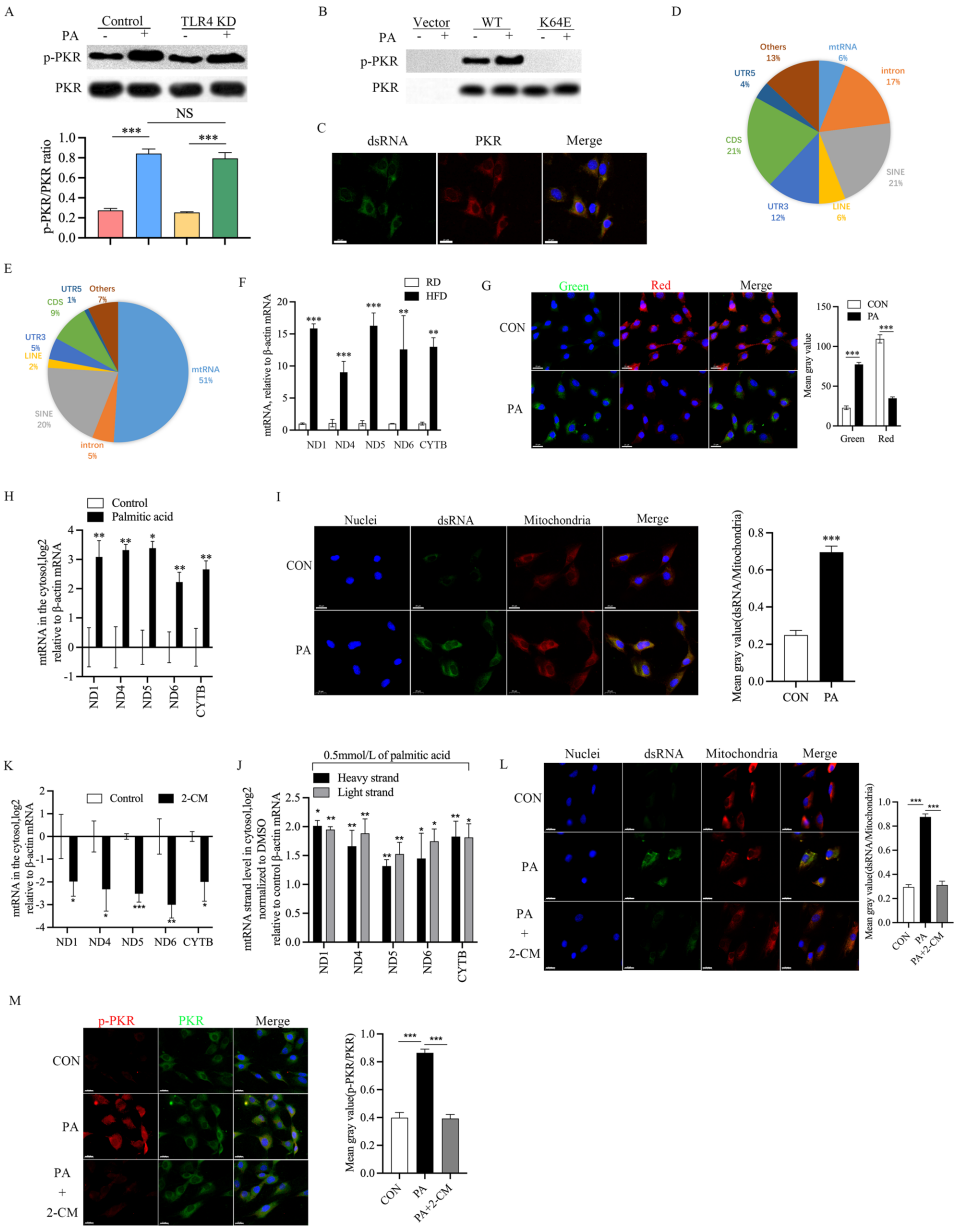
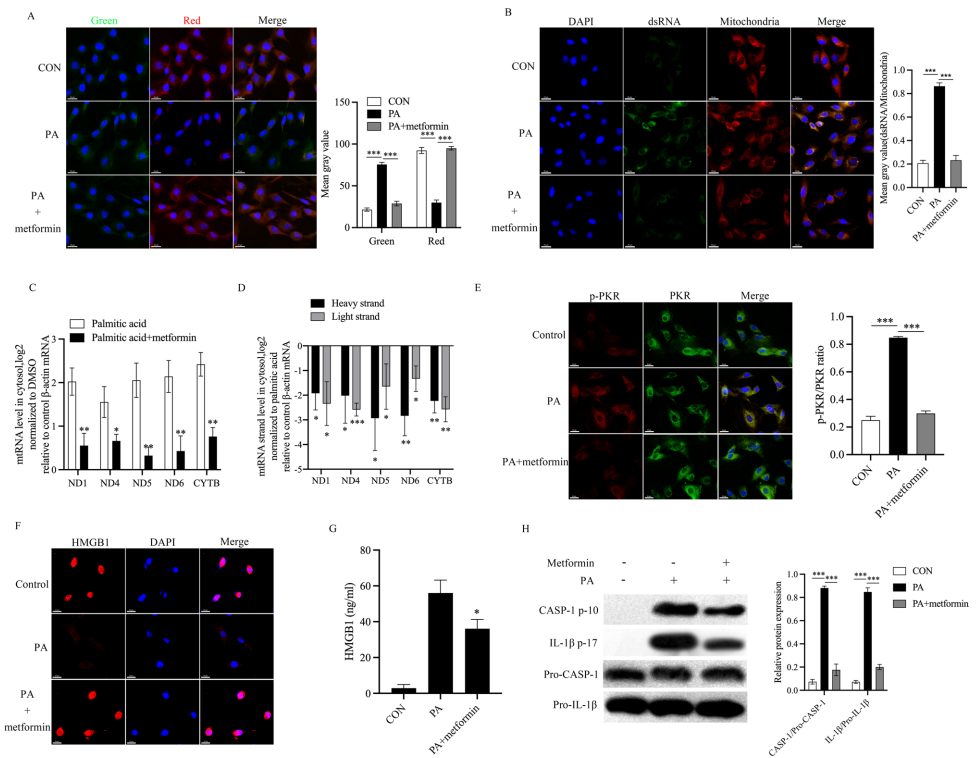
 p<0.05,*p<0.01 by Student's t test,n=3,immunohistochemistry of p-PKR and PKR on the NP tissue of control, IVDD and metformin treatment(limit or delay) group,scale bar=20μm,n=3,J immunofluorescence staining for IL-1β(red), caspase-1(red) and GSDMD-N(red) in the IVD degeneration model mice treated with metformin(limit or delay).Nucleus(blue),scale bar=50μm,n=3,Kimmunofluorescence staining for collagen-II(red),aggrecan(red),MMP-13(red) and ADAMTS-5(red) in the IVD degeneration model mice treated with metformin(limit or delay).Nucleus(blue),scale bar=100μm,n=3'>
p<0.05,*p<0.01 by Student's t test,n=3,immunohistochemistry of p-PKR and PKR on the NP tissue of control, IVDD and metformin treatment(limit or delay) group,scale bar=20μm,n=3,J immunofluorescence staining for IL-1β(red), caspase-1(red) and GSDMD-N(red) in the IVD degeneration model mice treated with metformin(limit or delay).Nucleus(blue),scale bar=50μm,n=3,Kimmunofluorescence staining for collagen-II(red),aggrecan(red),MMP-13(red) and ADAMTS-5(red) in the IVD degeneration model mice treated with metformin(limit or delay).Nucleus(blue),scale bar=100μm,n=3'>生物通 版权所有





