编辑推荐:
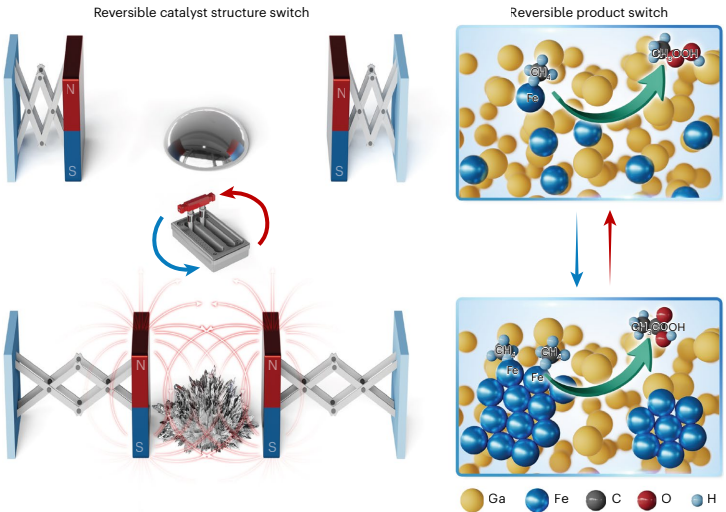
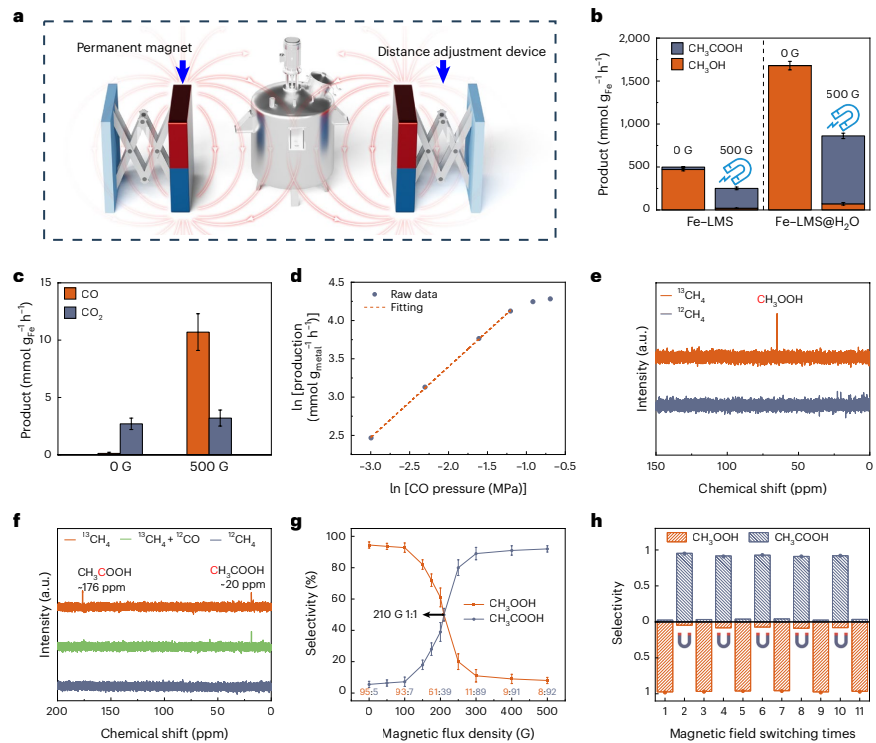
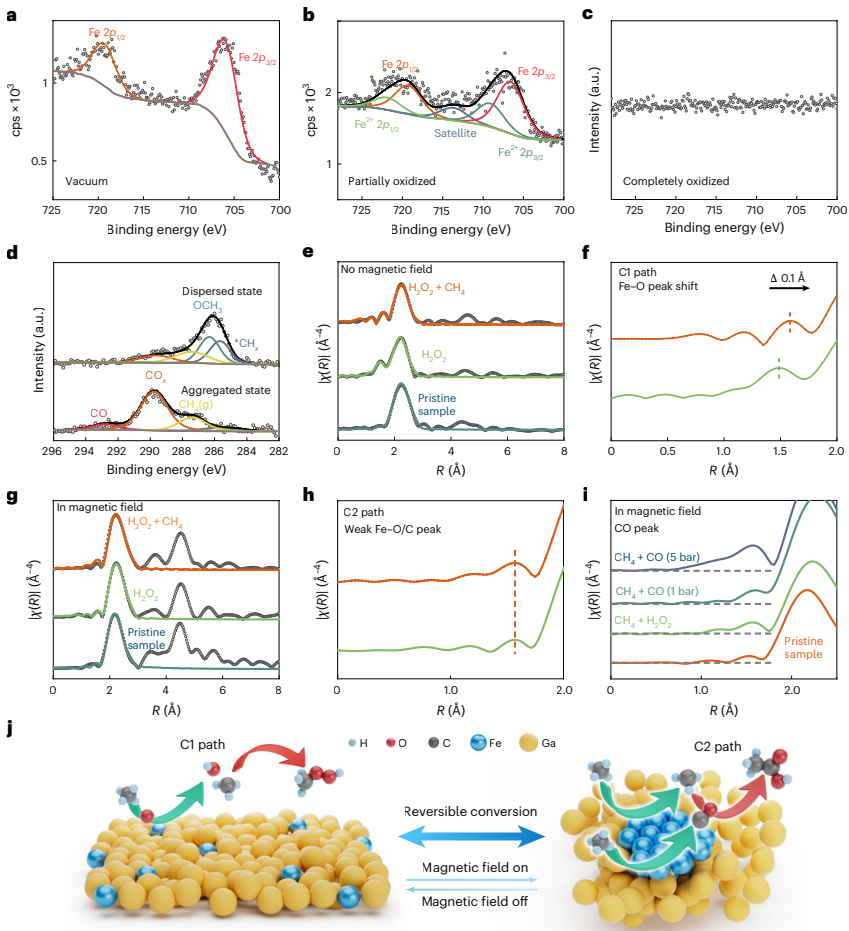
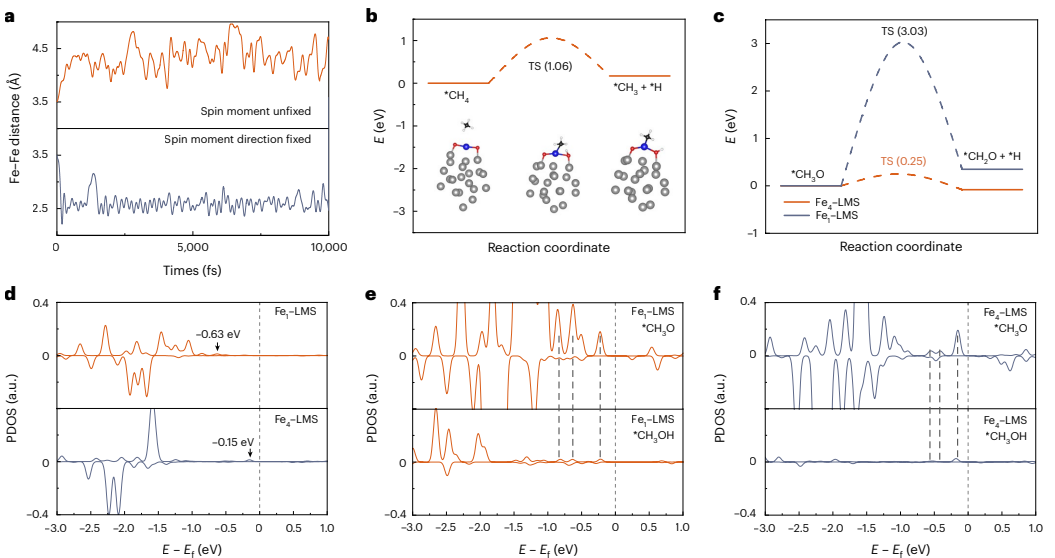
本研究针对传统甲烷氧化催化剂活性位点结构刚性、难以动态调控的问题,开发了一种铁嵌入液态金属催化剂(Fe-LMS),通过外磁场控制铁原子的聚集状态和自旋取向,实现了甲烷氧化主要液态产物在甲基过氧化物(CH3OOH)和乙酸(CH3COOH)之间的可逆转换。该催化剂在室温条件下展现出优异的生产速率(CH3OOH: 1,679.6 mmol gFe-1h-1;CH3COOH: 790.5 mmol gFe-1h-1)和选择性(分别达99.9%和91.7%),为精准控制催化反应路径提供了新策略。
生物通 版权所有





