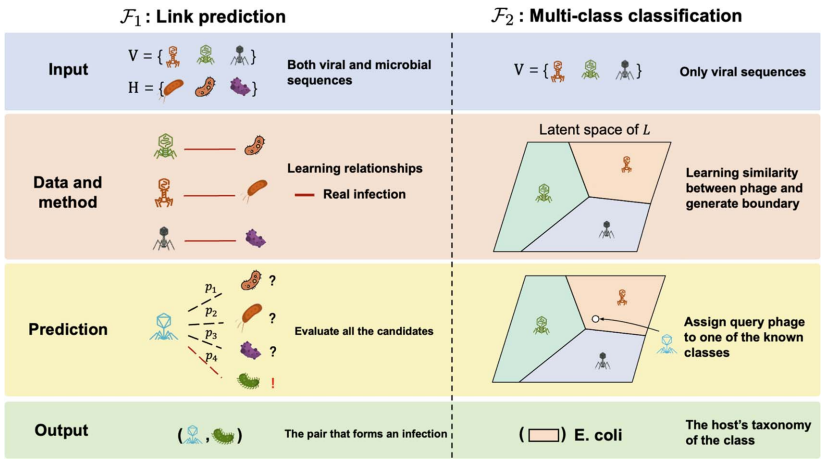
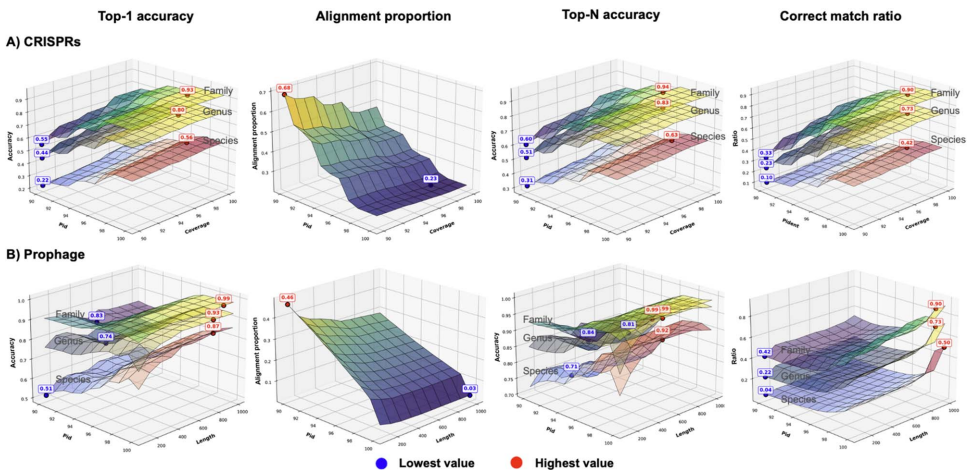
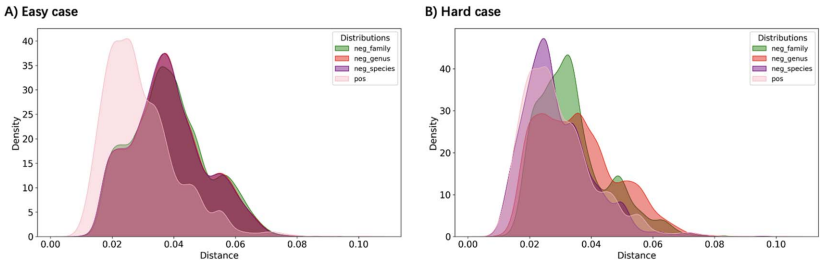
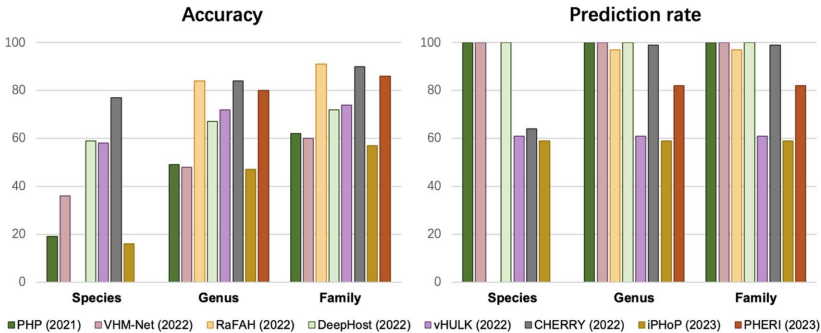
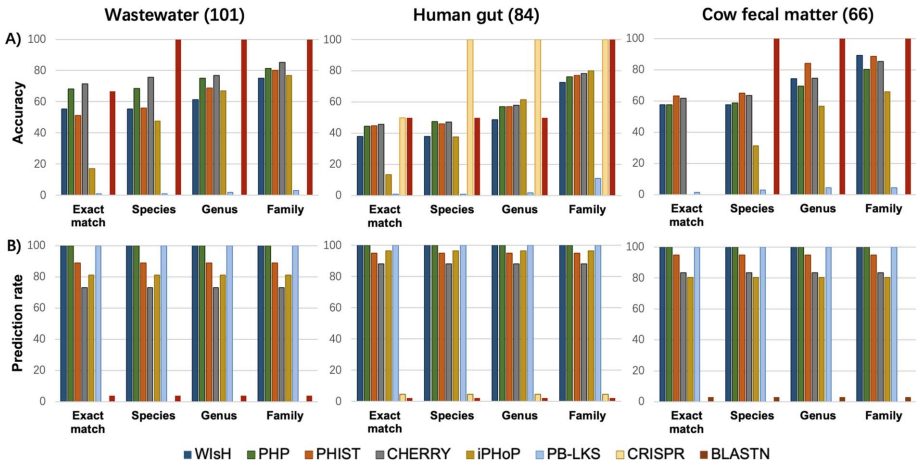
在微生物世界的隐秘战争中,噬菌体作为地球上最丰富的生物实体,与细菌宿主上演着永不停息的进化军备竞赛。这些病毒与宿主的相互作用不仅塑造着微生物群落的生态结构,更是抗感染治疗的新希望——噬菌体疗法正成为对抗抗生素耐药性的有力武器。然而,实验鉴定这些相互作用既耗时又受限于宿主培养要求,随着高通量测序技术的爆发式发展,计算预测方法应运而生。但问题随之而来:越来越多的预测工具构成了一个复杂的技术生态,由于评估标准不一致和工具可用性差异,直接比较它们的性能变得异常困难。更棘手的是,现有数据库存在严重的注释缺口和研究偏见,大量病毒序列缺乏宿主信息,而已知的相互作用又高度集中在少数模式生物上。此外,病毒宿主范围的实际复杂性远超数据库简化的“一对一”关系,许多噬菌体能够感染多个物种甚至跨属传播,这给预测模型的构建带来了巨大挑战。为了厘清这一混乱局面,香港城市大学和香港中文大学的研究团队在《Briefings in Bioinformatics》上发表了重磅研究,对27种病毒-宿主预测工具进行了系统梳理和严格评估。他们首先将宿主预测任务明确划分为两大框架:链接预测(link prediction)和多类分类(multi-class classification),并构建了两个专门设计的基准数据集——RefSeq-VHDB(数据库中心型)和MetaHiC-VHDB(宏基因组发现型),以模拟不同的研究场景。