编辑推荐:
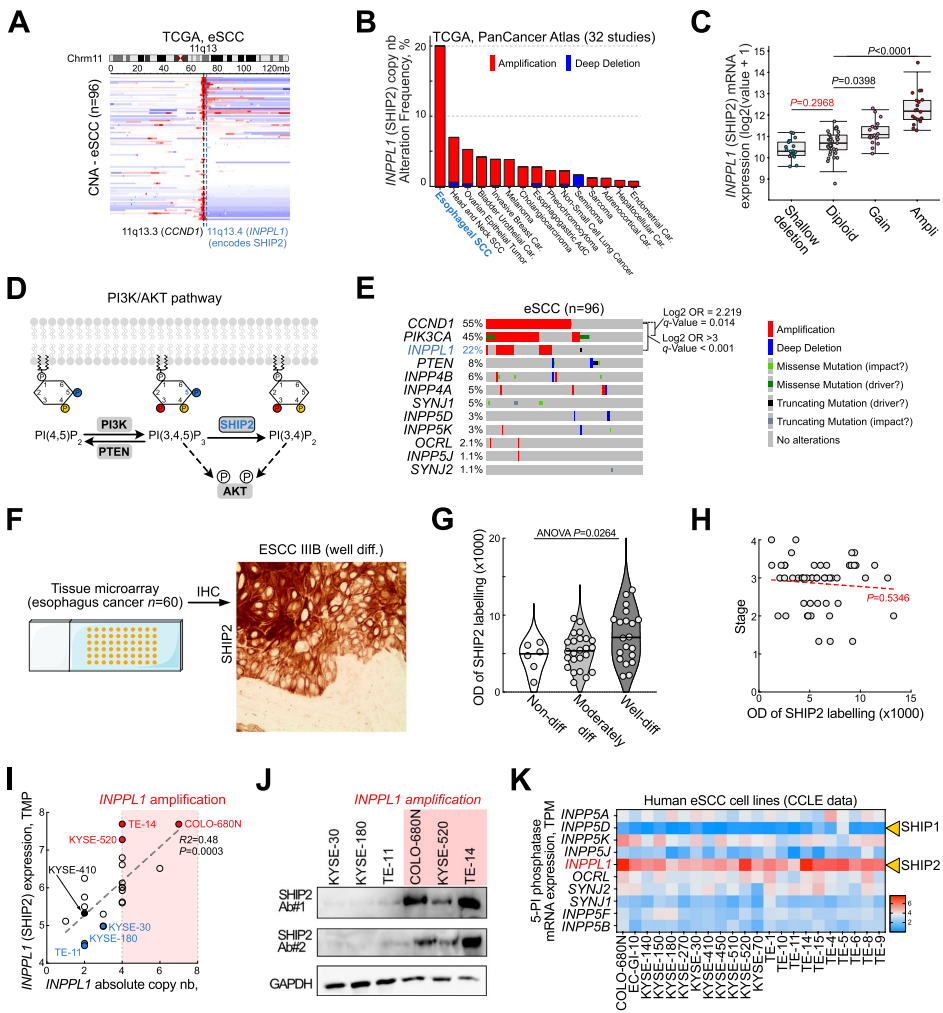
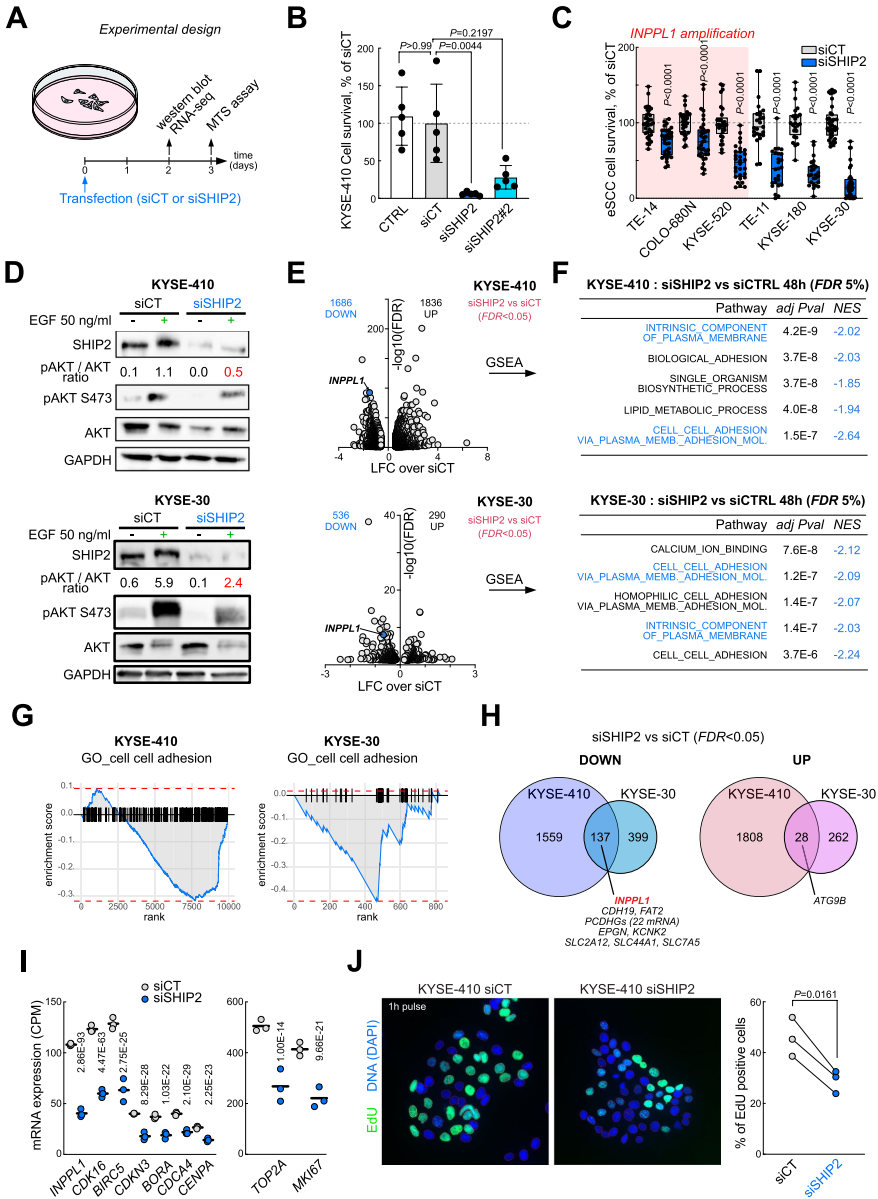
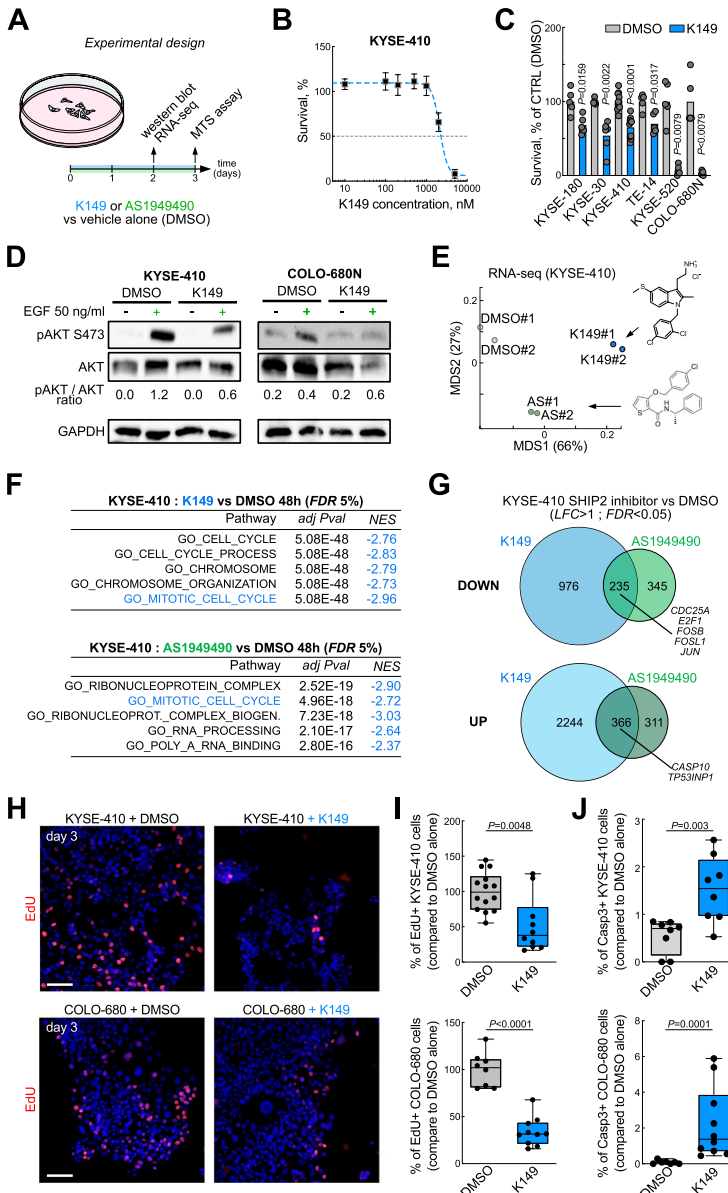
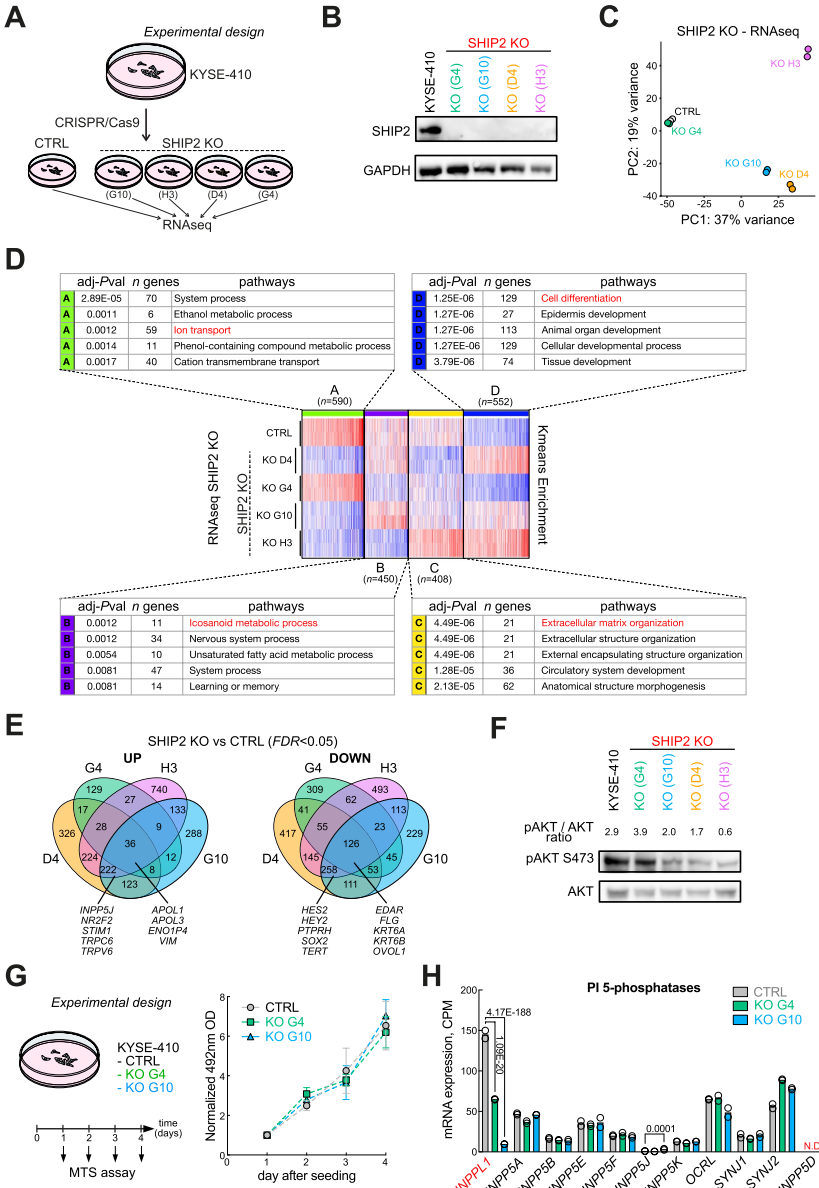
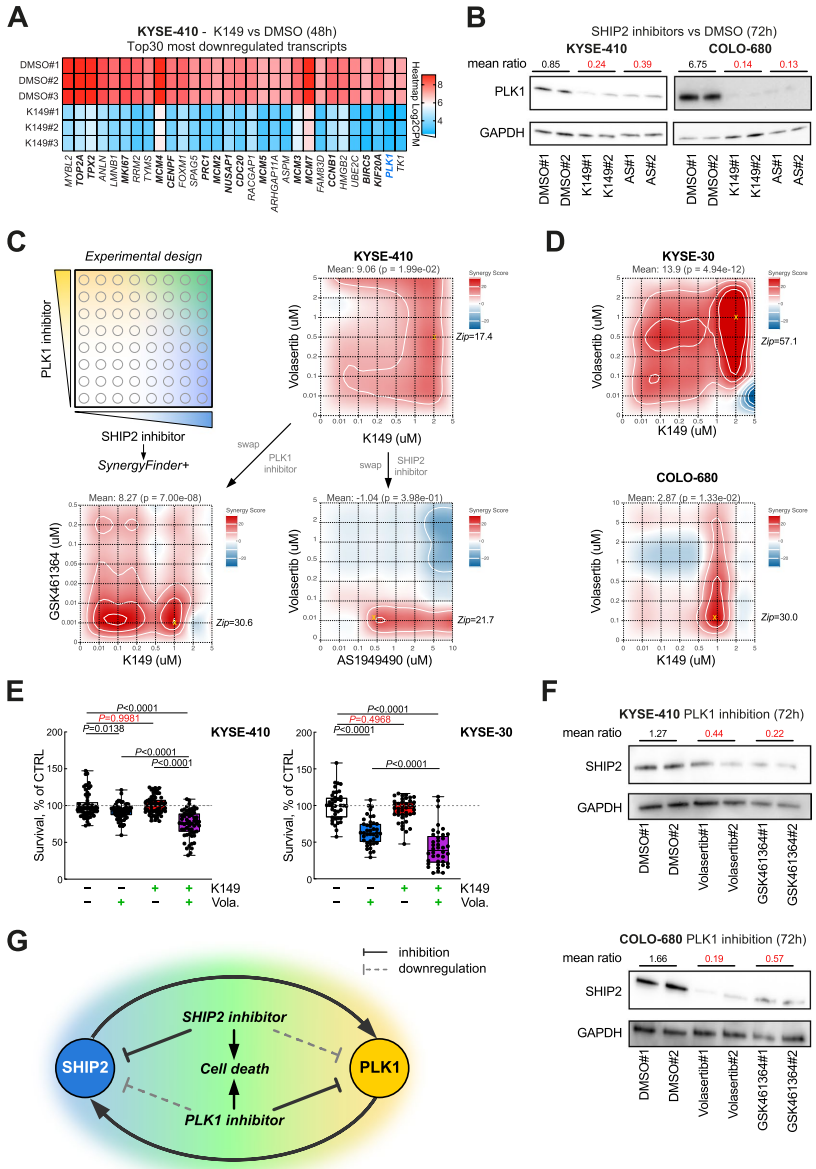
本研究针对食管鳞状细胞癌(eSCC)治疗耐药难题,发现INPPL1基因扩增导致SHIP2过表达并通过PI3K/AKT通路促进肿瘤生长。研究人员通过SHIP2敲降和药物抑制实验,证实其调控eSCC细胞存活/增殖的关键作用,并首次揭示SHIP2抑制可下调PLK1表达,双重抑制SHIP2与PLK1产生显著协同效应,为eSCC靶向治疗提供新策略。
 生物通微信公众号
生物通微信公众号
生物通 版权所有






